
- Scale of Mutation: Single Nucleotide Variations, Small-Scale Mutations, Structural Variations, Chromosomal Alterations
- Effect on DNA Sequence: Point Mutations, Insertions and Deletions, Copy Number Variations, Large-Scale Chromosomal Alterations
- Effect on Gene Product: Synonymous Mutations, Non-synonymous Mutations, Regulatory Mutations, Loss-of-Function Mutations, Gain-of-Function Mutations
- Location in the Genome: Coding Region Mutations, Non-coding Region Mutations
- Origin of Mutation: Germline Mutations, Somatic Mutations
- Inheritance Patterns: Dominant, Recessive, X-linked and Y-linked Mutations, Mitochondrial Inheritance
- Frequency and Distribution: Private Mutations, Common Variants
- Impact on the Organism: Benign Mutations, Pathogenic Mutations, Conditional Mutations
The Complexity of Genomic Disorders: Understanding the Challenges
The Diversity of Genomic Disorders

Prerequisites: None.
Level: Beginner.
Learning objectives:
Learning objectives:
- Grasp the range of genetic variations and their potential to cause genomic disorders.
- Understand the diversity of genomic disorders and their distinction from monogenic diseases.
- Recognize the role of environmental factors in the manifestation and progression of genomic disorders.
- Appreciate the importance of a comprehensive approach to studying genomic disorders, considering both genetic underpinnings and environmental influences.
Studying genomic disorders in exploring human health and disease presents a fascinating yet intellectually stimulating landscape. These disorders are defined by a broad spectrum of conditions resulting from variations within the human genome. Unlike monogenic diseases, which are attributed to mutations in a single gene, genomic disorders encompass a wide array of genetic alterations. These range from minimal changes in the nucleotide sequence to significant structural genomic rearrangements, including insertions, deletions, duplications, and complex chromosomal reconfigurations. This diversity underscores the complexity of genomic disorders, setting them apart as a unique and intellectually challenging area of study.
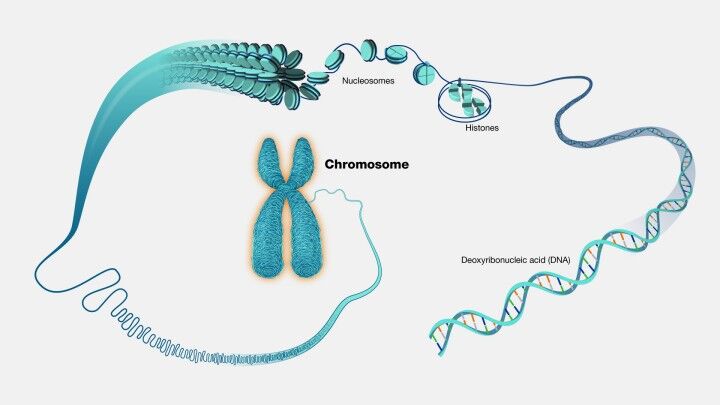
Understanding genomic disorders necessitates a comprehensive grasp of genetic variations. Such variations can manifest as minor alterations in the DNA sequence or as extensive changes affecting chromosomal structure (Figure 1) and the copy number of genes. These genetic anomalies play a critical role in gene expression and the synthesis and function of proteins within the organism. Although individual variations may not directly cause disease, a confluence of multiple variations, potentially interacting in intricate ways, can predispose an individual to various genomic disorders. This interplay of genetic and sometimes environmental factors contributes to the multifaceted nature of these conditions.
The implications of genomic disorders extend far beyond the cellular level, affecting an individual's overall physiology and health. Given the wide range of possible genetic modifications, these disorders can impact any part of the human genetic makeup, influencing the function and regulation of numerous genes. This variability in genetic impact leads to diverse symptoms and severity among affected individuals, depending on the specific genes involved and the magnitude of the genomic alteration. Consequently, diagnosing and treating genomic disorders presents significant challenges, necessitating a personalized approach to understanding each individual's genetic makeup.
Moreover, the development and progression of genomic disorders are not solely governed by genetic factors; environmental influences also play a crucial role. External factors, such as exposure to toxins, lifestyle choices, stress levels, and even the prenatal environment, can significantly affect gene expression and function. These environmental factors can act upon genetic susceptibilities, triggering or exacerbating the manifestation of diseases in individuals predisposed to certain conditions. For instance, a genetically predisposed individual may only exhibit symptoms of a condition like type 2 diabetes when coupled with environmental risk factors such as an unhealthy diet or insufficient physical activity.
The dynamic interplay between genetic and environmental factors highlights the complexity of genomic disorders. This interaction is not static but evolves, influencing an individual's risk of developing these conditions under varying environmental circumstances. Renowned examples of genomic disorders, such as DiGeorge Syndrome , Williams Syndrome, and Charcot-Marie-Tooth Disease Type 1A, illustrate these conditions' diverse manifestations and challenges. Understanding the intricate web of genetic and environmental factors that contribute to genomic disorders is crucial for devising effective prevention, diagnosis, and treatment strategies. This endeavor demands a holistic approach, integrating insights into the genetic foundation of these disorders with a nuanced understanding of the environmental contexts in which they occur.
Understanding Genetic Variation Through Classification

Delving into the realm of genetic variations requires a meticulous approach to understanding their nature, consequences, and implications for human health and disease. Classifying genetic mutations into distinct categories facilitates a structured exploration of their diverse effects on DNA sequences, the proteins they encode, their occurrence in either germ or somatic cells, and their patterns of inheritance. This systematic classification serves as a cornerstone for navigating the intricate world of genetic mutations, highlighting their complex and multifaceted impact on biological functions.
The framework for classifying genetic mutations is broad and encompasses various dimensions, each shedding light on different aspects of genetic variation. However, it is essential to acknowledge that the boundaries between these categories are not always sharply defined. This fuzziness mirrors the complexity of genetic mutations, which often transcend simple categorization due to their interconnected and overlapping nature.
Firstly, mutations can be classified based on the scale of the alteration they represent, encompassing single nucleotide variations (SNVs), small-scale mutations, structural variations, and chromosomal alterations. Such a classification allows for a granular understanding of the mutation's scope, from the smallest change in a single DNA building block to large-scale chromosomal rearrangements.
Another dimension considers the mutation's impact on the DNA sequence, distinguishing among point mutations, insertions and deletions (indels), copy number variations (CNVs), and extensive chromosomal modifications. This classification sheds light on the specific nature of the genetic alteration and its potential to disrupt normal genetic function.
Furthermore, mutations are categorized based on their effect on the gene product, including synonymous mutations that do not alter the amino acid sequence of proteins, non-synonymous mutations that do, regulatory mutations affecting gene expression, and loss-of-function or gain-of-function mutations that respectively diminish or enhance protein function. This classification underscores the direct implications of mutations on protein synthesis and function.
The location of mutations within the genome also serves as a basis for classification, distinguishing between mutations in coding regions, which affect protein-encoding DNA sequences, and those in non-coding regions, which can influence gene expression and regulation without altering protein sequences.
Additionally, the origin of mutation provides a critical classification criterion, differentiating between germline mutations, which are heritable and affect all cells of an organism, and somatic mutations, which occur in non-reproductive cells and are not passed to offspring. This distinction is vital for understanding specific mutations' heritability and potential generational impact.
Inheritance patterns further categorize mutations into dominant, recessive, X-linked, Y-linked, and mitochondrial, each with unique implications for genetic disease manifestation and transmission.
The frequency and distribution of mutations within populations also form a basis for classification, separating private mutations found in individual families from common variants present across broader populations. This distinction helps elucidate the role of specific mutations in genetic diversity and disease predisposition.
Lastly, the classification based on impact on the organism distinguishes between benign mutations, which do not significantly affect the organism's phenotype or fitness; pathogenic mutations, which lead to disease; and conditional mutations, whose effects depend on environmental factors. This categorization highlights the functional consequences of genetic variations, underscoring the delicate balance between mutation, health, and disease.
This multifaceted classification system gives us invaluable insights into the complex world of genetic variations. Understanding these categories illuminates the biological mechanisms underlying genetic disorders. It guides the development of targeted diagnostic and therapeutic strategies, paving the way for personalized medicine and a deeper comprehension of human genetics.
Synonymous Mutations
In the expansive field of genetics, synonymous mutations represent a nuanced aspect that underscores the intricate relationship between DNA sequences and protein synthesis. These mutations, often called silent mutations, testify to the redundancy inherent in the genetic code. Despite changes in the DNA sequence, the amino acid sequence of the resultant protein remains unaltered. This intriguing phenomenon is primarily due to the genetic code's degeneracy, wherein multiple codons can encode a single amino acid. An illustrative example of this redundancy can be observed in the encoding of the amino acid leucine, which can be represented by six different codons—UUA, UUG, CUU, CUC, CUA, and CUG in RNA or their DNA equivalents (Figure 1, Figure 2). This critical concept ties closely to the principles of codon usage, a foundational aspect of genetic coding and expression detailed further in Box "Codon Usage and Genetic Code Redundancy."
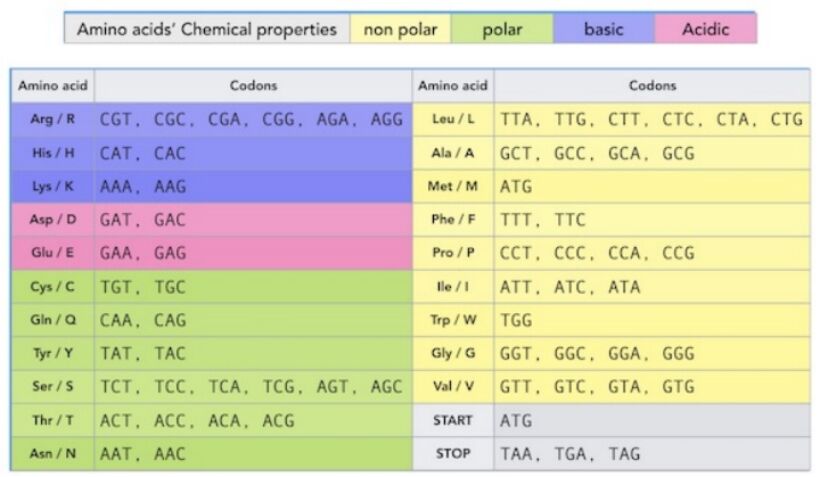
Synonymous mutations, despite not altering the amino acid sequence and thus the protein's structure and function, have significant biological and evolutionary implications (Figure 3). One of the key considerations arising from such mutations is the phenomenon of codon usage bias. This aspect refers to the preference of certain codons over others for the same amino acid across different organisms, a factor that can influence the efficiency and fidelity of protein translation. Such biases can lead to variations in gene expression levels, underscoring the evolutionary significance of synonymous mutations.
Beyond the scope of codon preference, synonymous mutations exert their influence on RNA dynamics as well. Although the protein's amino acid sequence remains constant, alterations in the RNA sequence can affect RNA stability, structure, and processing mechanisms. These changes, in turn, can indirectly modulate the functionality and abundance of the corresponding protein, revealing the layered complexities synonymous with mutations introduced at the molecular level.
Furthermore, the potential regulatory implications of synonymous mutations extend to gene splicing (Figure 2), the functionality of regulatory elements, and microRNA (miRNA) binding sites. By influencing these critical regulatory pathways, synonymous mutations can precipitate gene expression and functionality changes, even without directly altering the protein's amino acid sequence, underscores the broader impacts of synonymous mutations, highlighting their capacity to influence genetic expression and function through a variety of molecular mechanisms, thereby contributing to the ongoing evolution and adaptation of organisms at the genetic level.
Studying synonymous mutations offers profound insights into genetic coding, expression, and regulation complexity. It illuminates the nuanced ways the genetic code can be rigid in encoding proteins and flexible in its capacity for variation and adaptation. As such, synonymous mutations are a pivotal area of study within genetics and bioinformatics, offering a window into the delicate balance between genetic stability and variability that underpins life.
Codon Usage Bias: Variability and Influence
Codons, sequences of three nucleotides, are the basic units of the genetic code, mapping to amino acids or stop signals during protein synthesis. The redundancy in the genetic code, with 64 codons but only 20 amino acids, leads to synonymous codons and introduces the phenomenon of codon usage bias.
This bias refers to the preference for certain codons over others, varying across organisms and influenced by factors like genomic composition and tRNA abundance. Understanding these preferences is crucial for optimizing gene expression and protein synthesis.
Factors Influencing Codon Usage
- Genomic Nucleotide Composition: Preference can be affected by the genome's GC or AT richness, particularly at the wobble position.
- tRNA Abundance: Cells may prefer codons matching their most abundant tRNAs, enhancing translation efficiency.
- Translation Efficiency and Accuracy: Certain codons may be selected for their role in reducing translational errors.
The Wobble Position
The flexibility at the third codon position allows a single tRNA to recognize multiple codons, influencing codon preference and efficiency in protein synthesis.
Impact of Codon Usage Bias
Codon usage bias affects protein levels, folding, and function, highlighting its significance in genetic engineering and synthetic biology for optimizing gene synthesis and expression.
Non-synonymous Mutations
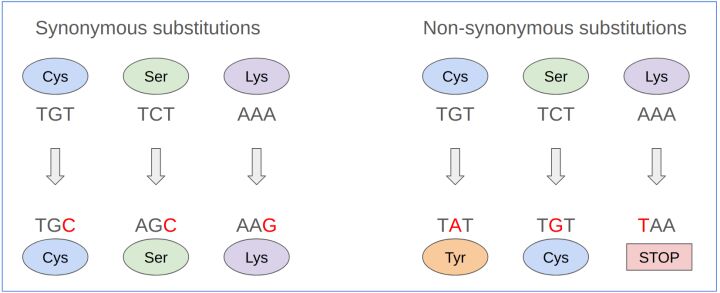
Non-synonymous mutations occupy a pivotal role due to their direct influence on the amino acid sequence of proteins, potentially altering their function. These mutations contrast with synonymous mutations, which do not affect the amino acid sequence despite changes in the DNA (Figure 3). Non-synonymous mutations can be categorized into three main types: missense, nonsense, and readthrough mutations, each with distinct implications for protein structure and function.
Missense mutations involve the substitution of one amino acid for another within the protein sequence. The impact of such mutations on the protein's function varies widely, influenced by the specific amino acids involved and the location of the change within the protein. Some substitutions may have minimal effects, preserving protein function, while others can drastically alter the protein's properties or disrupt its function entirely.
Nonsense mutations represent a significant alteration where a codon encoding an amino acid is changed to a stop codon. This change results in premature termination of translation, leading to a truncated protein that often lacks functional capability. These truncated proteins are typically subject to rapid degradation, further diminishing their potential functional contributions within the cell.
Readthrough mutations occur when a stop codon is altered to encode an amino acid, thereby extending the protein's length beyond its original endpoint. This elongation can interfere with normal protein function and stability, as the additional amino acid residues may disrupt the protein's three-dimensional structure or its interactions with other molecules.
The significance of non-synonymous mutations extends beyond their immediate effects on protein function to broader implications for genetic diversity, disease pathogenesis, and evolutionary processes. These mutations can alter the structural configuration, stability, or interaction capabilities of proteins, potentially leading to either a loss or gain of function. The study of non-synonymous mutations is therefore crucial for understanding the molecular mechanisms underlying various diseases, particularly those that compromise protein functionality.
Moreover, non-synonymous mutations contribute to the diversification of life and the evolutionary adaptation of protein functions. Natural selection acts upon these mutations, preserving beneficial alterations that enhance organismal fitness while eliminating or reducing the frequency of deleterious changes.
In contrast to non-synonymous mutations, regulatory mutations do not alter the amino acid sequence of proteins. Instead, they affect the regulation of gene expression, including the timing, location, and level of gene product synthesis. Such mutations can occur in several genomic regions responsible for gene expression control, such as promoters, enhancers, silencers, and elements involved in RNA processing. The distinction between regulatory and non-synonymous mutations underscores the multifaceted nature of genetic variation and its complex influence on organismal phenotype, disease susceptibility, and evolutionary trajectory.
Understanding both non-synonymous and regulatory mutations is fundamental to elucidating the genetic underpinnings of physiological processes, disease development, and the evolutionary dynamics shaping biological diversity. This comprehension aids in the development of targeted therapeutic strategies and enhances our grasp of the genetic basis of adaptation and speciation.
Mutations in Regulatory Regions - Types of Regulatory Mutations
Genetic regulation requires a detailed understanding of mutations that occur within regulatory regions. These mutations play a crucial role in the expression of genes and can significantly impact an organism's phenotype. Furthermore, mutations in DNA sequences that are critical for controlling gene activity can lead to a variety of diseases. This exploration aims to shed light on the different types of regulatory mutations and their impact on biological functions and evolutionary processes.
Regulatory mutations in promoter regions, which serve as crucial binding sites for RNA polymerase and other transcription factors, are central to the initiation of gene transcription (Figure 4). Alterations within these sequences can significantly modulate gene expression levels, either by enhancing or suppressing the transcription process. Such mutations, by affecting the rate at which genes are expressed, can lead to a range of phenotypic outcomes depending on the nature and extent of the expression change.
Furthermore, mutations in enhancer and silencer regions underscore the delicate balance of gene expression. Enhancers, through their interaction with specific transcription factors, augment the transcription levels of genes they regulate, whereas silencers perform the inverse function, repressing gene activity. Mutations in these regions can disrupt the intricate regulation of gene expression, leading to an imbalance that may contribute to pathological conditions.
The integrity of splicing sites is critical for the accurate processing of pre-mRNA, specifically the excision of introns and the ligation of exons. Mutations in these regions can result in aberrant splicing, potentially leading to the production of dysfunctional proteins or the degradation of mRNA. Such errors in mRNA processing underscore the importance of precise splicing in protein synthesis and the potential consequences of its disruption.
Mutations within the 5' and 3' untranslated regions (UTRs) of mRNAs also play a significant role in post-transcriptional regulation, influencing mRNA stability, localization within the cell, and the efficiency of translation initiation. These regions, therefore, are crucial in determining the levels of protein synthesized within the cell, with mutations potentially leading to varied phenotypic effects.
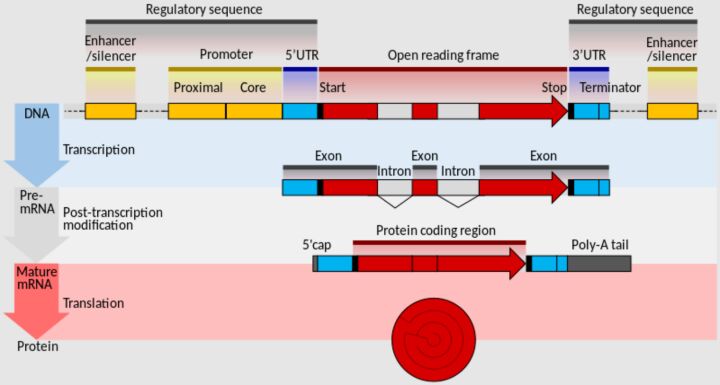
Additionally, the role of microRNA (miRNA) target sites in regulating gene expression highlights the complexity of post-transcriptional regulation. miRNAs, by binding to complementary sequences within the 3' UTRs of their target mRNAs, can suppress gene expression through mRNA degradation or inhibition of translation. Mutations that disrupt or create new miRNA binding sites can thus significantly alter gene expression patterns, with implications for cellular function and disease progression.
Alterations in the sequences necessary for RNA editing further illustrate the multifaceted nature of genetic regulation. Given that RNA editing modifies the nucleotide sequence of an RNA molecule post-transcription, mutations in these sites can have profound effects on the RNA molecule's function, affecting the biological activity of the encoded protein.
The implications of regulatory mutations extend beyond mere phenotypic alterations. They can disrupt the precise gene expression patterns necessary for normal development, leading to congenital disorders, and contribute to the dysregulation of genes critical for cell growth, differentiation, and death, thereby playing a role in cancer development. The subtle yet significant changes in gene expression levels caused by these mutations also contribute to the variability observed among individuals with the same genetic condition.
On an evolutionary scale, regulatory mutations facilitate adaptation by allowing for changes in gene expression patterns without altering protein function. This capacity for regulatory flexibility enables organisms to adjust to new environmental challenges, highlighting the essential role of regulatory mutations in the evolution of species.
This comprehensive overview of regulatory mutations underscores the complexity of genetic regulation and its profound impact on biological systems. Understanding these mutations not only sheds light on the molecular mechanisms underlying various diseases but also provides insights into the evolutionary dynamics that shape the diversity of life.
Types and Impacts of Non-coding Region Mutations
In the vast and intricate genome landscape, mutations within non-coding regions—once relegated to the category of "junk DNA"—have emerged as significant factors influencing gene expression, genomic integrity, and cellular functions. These regions, devoid of protein-coding sequences, harbor elements crucial for regulating genes and maintaining genomic architecture. Understanding the types and impacts of mutations in these areas is essential for grasping the broader implications for organismal biology and disease.
Intergenic region mutations occur within the stretches of DNA that lie between genes. Far from being inert, these regions are rich with regulatory elements and non-coding RNAs that have pivotal roles in controlling gene expression and shaping the genomic landscape. Mutations in these areas can disrupt long-range gene regulation, affecting gene expression in complex ways extending beyond the mutation's immediate vicinity. Additionally, alterations in intergenic regions can influence the overall architecture of the genome, with potential consequences for chromosomal structure and integrity.
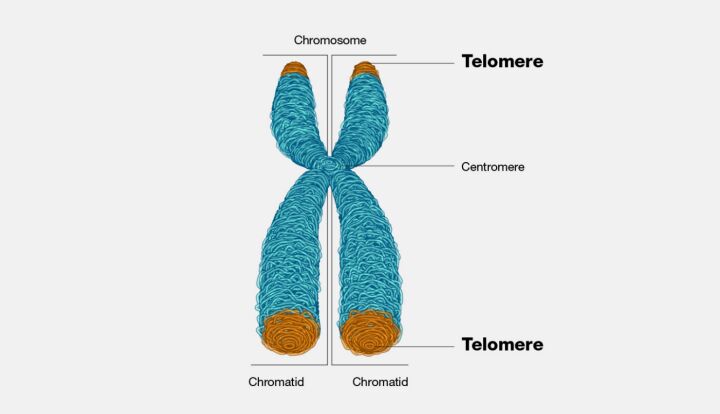
Mutations that influence the length of telomeres represent another category with profound biological implications. Telomeres, the protective caps at the ends of chromosomes, play a critical role in chromosome stability and cellular lifespan. Changes in telomere length, whether through deletion, duplication, or disruption of telomere maintenance mechanisms, can lead to chromosomal instability, a hallmark of cancer cells, and influence the process of cellular aging. These mutations have garnered interest for their potential connections to cancer development and age-related diseases, highlighting the importance of telomere integrity for cellular health and longevity (Figure 5).
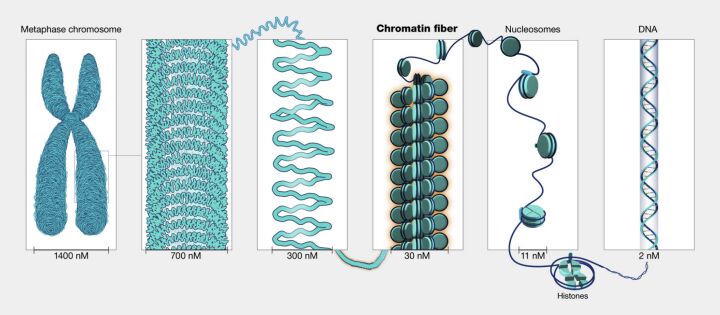
Another significant type of mutation affects regulatory elements involved in chromatin structure (Figure 6). Chromatin is a complex of DNA and proteins that makeup chromosomes. Its structure undergoes changes that regulate DNA accessibility to transcription machinery. Mutations that alter the DNA sequences recognized by proteins involved in chromatin remodeling can profoundly impact gene expression. By influencing how tightly or loosely DNA is packaged, these mutations can enhance or suppress the expression of genes across the genome, leading to widespread effects on cellular function and development.
The study of mutations in non-coding genome regions sheds light on the complexity of genetic regulation and the delicate balance required for normal cellular function. These mutations can have far-reaching effects, influencing individual genes and the overall behavior of cells and organisms. As research unravels the mysteries of the non-coding genome, it becomes increasingly clear that these regions play crucial roles in health, disease, and the evolutionary processes that shape life.
Loss-of-Function Mutations
Loss-of-function (LoF) mutations represent a critical concept in the study of genetics and molecular biology, as they are fundamental to understanding the alteration of protein function and its consequences on cellular and organismal levels. We can characterize such mutations by their capacity to diminish or entirely inhibit the function of the protein encoded by the gene in question. This inactivation can arise through various mechanisms, impacting different facets of gene expression or the protein's structural integrity. Typically, LoF mutations are recessive; that is, the manifestation of a phenotype necessitates the inheritance of two mutated gene copies. Nevertheless, the phenomenon of haploinsufficiency illustrates an exception, where a single copy of a mutated gene is sufficient to produce a phenotype due to inadequate protein functionality, highlighting the nuanced nature of gene expression and protein function within biological systems.
The mechanisms underlying LoF mutations are diverse, each affecting the gene-to-protein pathway in unique ways. Nonsense mutations introduce a premature stop codon within the coding sequence, resulting in a truncated protein that typically lacks functional capability. On the other hand, missense mutations involve substituting one amino acid for another, profoundly affecting the protein's structure or active site and potentially compromising its function. Frameshift mutations, caused by insertions or deletions that do not occur in multiples of three, alter the gene's reading frame, usually producing a nonfunctional protein, as the subsequent sequence is significantly disrupted. Splice site mutations impact the accurate removal of introns and the fusion of exons, which can lead to the production of an aberrant and dysfunctional protein. Lastly, regulatory mutations modify the level of gene expression, resulting in the production of insufficient quantities of the protein.
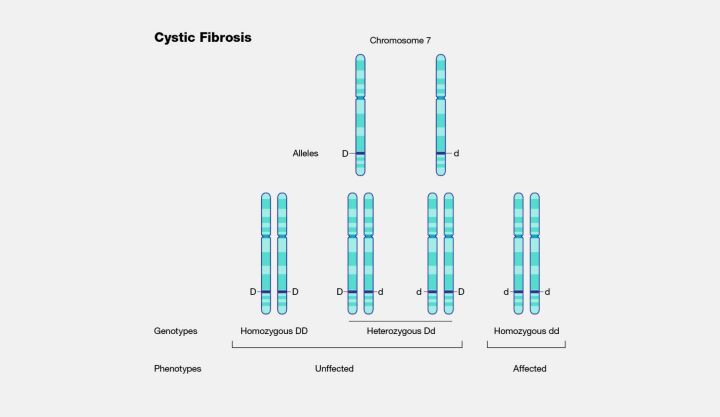
The implications of LoF mutations are vast and varied, with significant relevance to basic research and clinical applications. Many genetic disorders can be traced back to LoF mutations that reduce the functionality of crucial proteins. For instance, cystic fibrosis is a condition resulting from mutations in the CFTR gene, which impairs chloride ion transport (Figure 7). Beyond their role in elucidating the genetic basis of disease, LoF mutations also serve as a window into the molecular mechanisms underlying various conditions, offering potential targets for therapeutic intervention. Researchers can gain insights into disease mechanisms and identify novel treatment approaches by exploring the function of proteins rendered inactive by such mutations. Thus, the study of LoF mutations is fundamental for understanding genetic disease and the development of innovative therapeutic strategies, illustrating the profound impact of genetics on medical science.
Gain-of-Function Mutations
Gain-of-function (GoF) mutations represent a fascinating category of genetic alterations, as they imbue the protein product with new capabilities, enhanced functionalities, or atypical expression patterns that diverge from the norm established by the wild-type (or natural) phenotype. Unlike loss-of-function mutations, which typically diminish protein activity, GoF mutations can have an augmented biological effect. These mutations often exhibit a dominant inheritance pattern, where a single allele carrying the mutation is sufficient to alter the phenotype. This characteristic underscores the potent nature of GoF mutations in influencing organismal traits and disease phenologies.
Several mechanisms underpin the genesis of GoF mutations. Missense mutations, which alter the protein's amino acid sequence, can lead to an enhanced function or bestow a novel activity upon the protein. Another route to gain function is gene duplication, where an increase in the copy number of a gene leads to its overexpression, thereby amplifying its activity within the cell. Regulatory mutations can also play a role by boosting a gene's expression levels beyond typical norms or initiating expression in cell types or developmental stages where the gene is normally silent. Additionally, fusion proteins, born from translocations that amalgamate parts of two different genes, can create proteins with entirely new functions. Such mechanisms are especially relevant in the context of oncogenesis, where aberrant fusion proteins can drive the development of cancer.
The implications of GoF mutations are vast and intensely significant for understanding human disease and biology. In oncology, GoF mutations in oncogenes (Figure 8) are a common motif, leading to the unregulated proliferation and division of cells—a cornerstone of cancer development. Beyond cancer, these mutations can interfere with normal developmental processes, resulting in a spectrum of congenital anomalies. Furthermore, identifying GoF mutations within disease pathways offers a valuable target for therapeutic intervention, with drugs designed to inhibit the aberrant protein function providing a route to treat or manage disease conditions.
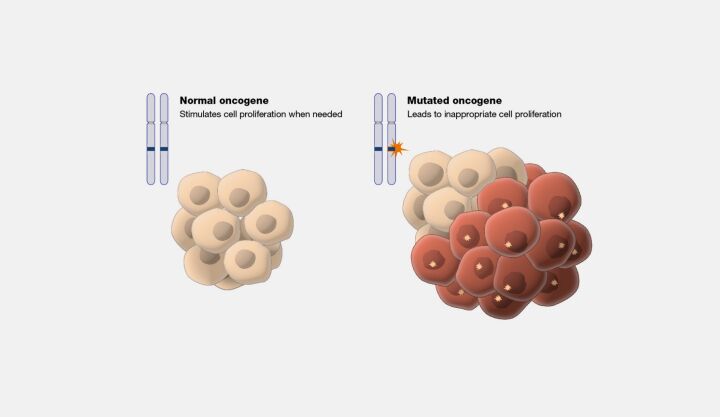
The study of GoF mutations, alongside loss-of-function mutations, enriches our comprehension of the intricate tapestry of genetic influence on biological systems. These mutations illuminate the pathogenesis of various diseases and offer insights into the dynamic processes of evolution and development. By decoding the complexities of GoF mutations, researchers and clinicians can unlock new avenues for diagnosing, treating, and understanding genetic disorders, marking a critical step forward in pursuing precision medicine and advanced therapeutic strategies.
Single Nucleotide Variations (SNVs)
Single Nucleotide Variations (SNVs) constitute the most fundamental form of genetic variation within organisms' genomes, underpinning the diversity observed across individuals. An SNV entails a modification in a single nucleotide—the foundational building blocks of DNA and RNA—altering the genomic sequence in a discrete yet potentially significant manner. The ubiquity of SNVs across the genome means they can manifest in coding regions, where they may influence protein structure and function, and in non-coding regions, where their impact on gene regulation and chromosomal architecture can be profound. The significance of SNVs extends across various domains of biological research, from elucidating the molecular basis of genetic diseases to tracing the evolutionary history of species.
The biological ramifications of SNVs are manifold and contingent on the nature and context of the nucleotide change. Point mutations represent a primary category of SNVs, characterized by substituting one nucleotide for another (Figure 9). We can further divide into transitions, which entail the substitution of a purine for a purine or a pyrimidine for a pyrimidine, and transversions, which involve the exchange of a purine for a pyrimidine or vice versa (Figure 10). Transitions are more frequent than transversions within the genome, reflecting a bias that has implications for the mutation rate and patterns of genetic variation.
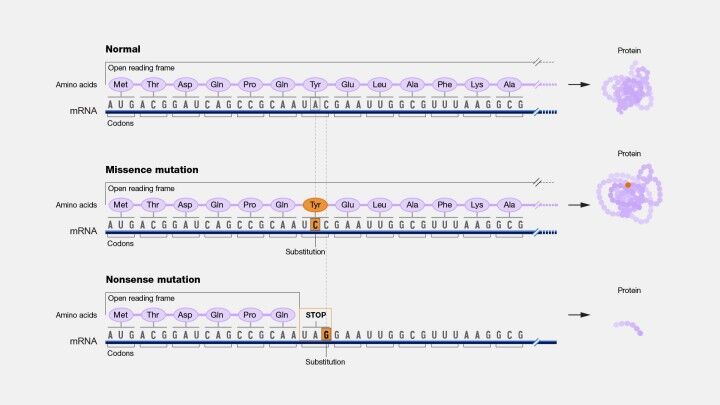
Among the consequences of SNVs, missense mutations are particularly noteworthy. These mutations alter the codon sequence such that a different amino acid is incorporated into the protein, potentially altering its functionality. The impact of a missense mutation is influenced by the nature of the amino acid substitution; changes that preserve the chemical properties of the original amino acid may have minimal effects, whereas substitutions that introduce chemically divergent amino acids can profoundly affect protein structure and activity. Missense mutations can have diverse outcomes, from benign variations to severe pathological conditions, exemplified by diseases like Sickle Cell Disease and Cystic Fibrosis, where single amino acid changes lead to significant physiological dysfunctions.
Nonsense mutations present another facet of SNVs, where the alteration results in a premature stop codon, truncating the protein product. The truncated protein, often lacking critical functional domains, is typically nonfunctional, leading to loss-of-function effects that can have deleterious consequences for the organism. The severity of the impact depends on various factors, including the position of the premature stop codon within the gene and the essentiality of the protein's function. Nonsense mutations are implicated in various genetic disorders, underscoring their significance in human health and disease.
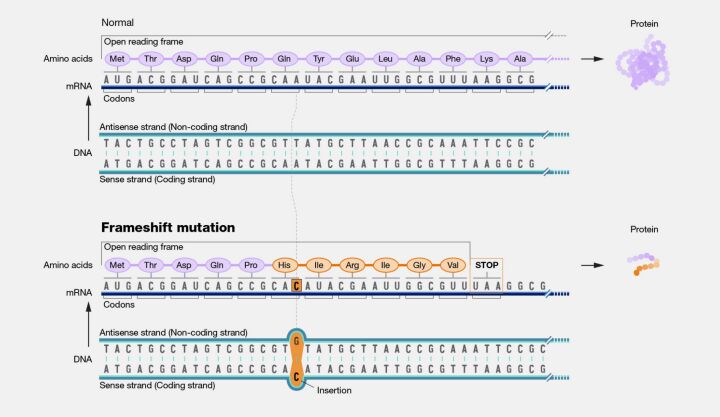
Frameshift mutations, although not strictly limited to single nucleotide changes, can result from SNVs that involve the insertion or deletion of nucleotides, leading to a shift in the reading frame of the genetic code (Figure 11). Such mutations can dramatically alter the downstream amino acid sequence, often resulting in nonfunctional proteins and severe phenotypic effects.
Exploring SNVs illuminates the intricate mechanisms by which genetic variation influences biological function and evolution. Understanding these variations is crucial for deciphering the genetic underpinnings of diseases, facilitating the development of diagnostic tools and therapeutic interventions, and advancing our comprehension of the genetic diversity that shapes life on Earth.
Small-Scale Mutations
Small-scale mutations are genetic alterations that affect a relatively small number of nucleotides within the genome. Typically, this scale ranges from a single nucleotide change (the most minor possible mutation) to mutations affecting a few dozen nucleotides. The exact upper limit of the size can vary depending on the context in which the term is used. However, it generally encompasses mutations limited to alterations within a single or a few genes rather than large segments of a chromosome or whole chromosomes.
These mutations can occur in various forms, including point, insertions, deletions, and frameshift mutations. They often result from errors in DNA replication or repair or mutagens' effects. Depending on their location and nature, small-scale mutations can have a wide range of effects on an organism, from benign to causing significant changes in protein function or expression, potentially leading to diseases.
Structural variations (SVs)
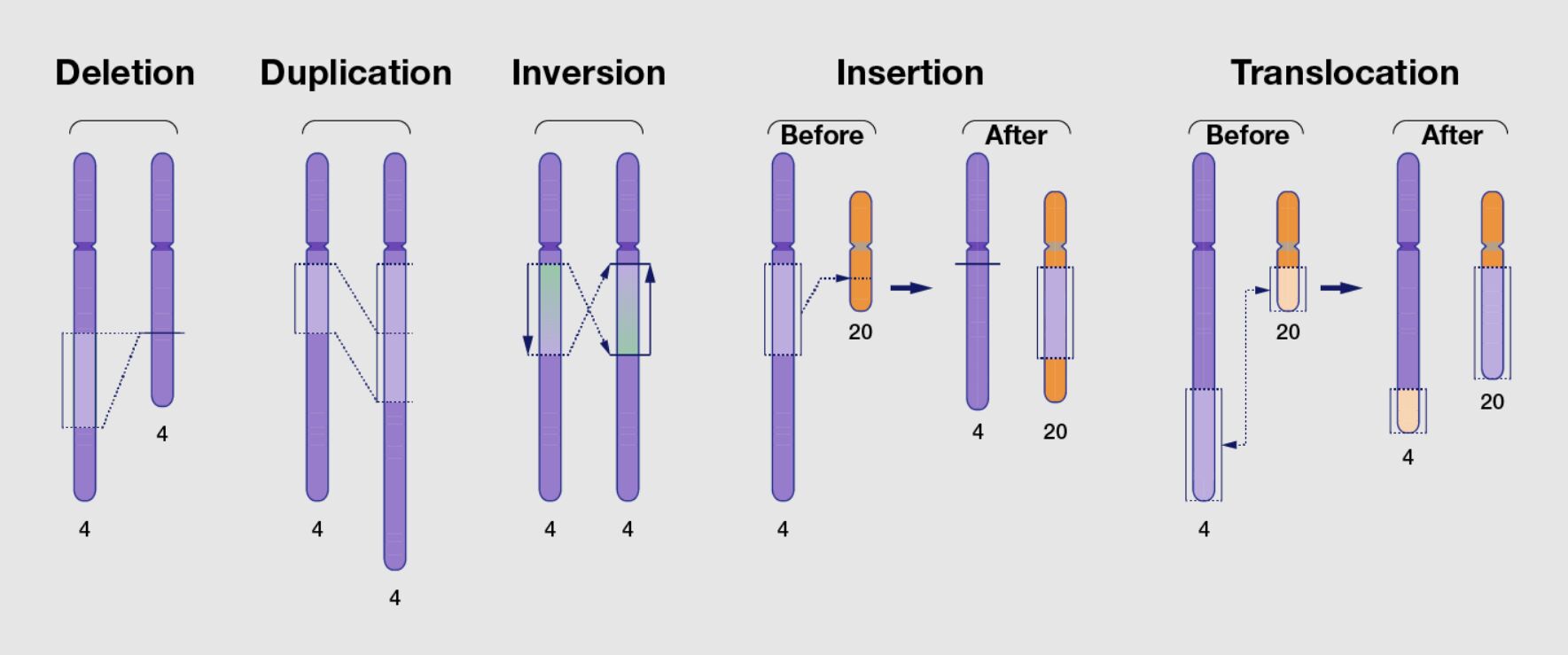
Structural variations (SVs) are large-scale genomic alterations that affect the structure of chromosomes and can significantly impact an organism's genome. Unlike small-scale mutations that involve changes to just a few nucleotides, structural variations involve longer DNA segments that are generally considered to be at least 1,000 base pairs (1 kilobase, kb) in size. However, this threshold can vary depending on the definition used by different researchers. SVs encompass a broad range of genomic rearrangements and can include:
Deletions are the loss of a segment of DNA from the chromosome. If critical genes are affected, this can result in the removal of one or more genes or regulatory elements, potentially leading to diseases.
Insertions are the addition of a segment of DNA to the chromosome. The insertion within a gene can disrupt gene function or add new genetic material to the genome.
Duplications. A genome segment is copied one or more times, increasing its copy number. If they disrupt normal gene function, duplications can contribute to genetic variability and evolution and cause disease.
Inversions. A segment of DNA is reversed end to end. If the inversion occurs within a gene or disrupts regulatory elements, it can affect gene function.
Translocations. A segment of DNA is moved from one location in the genome to another, which can be within the same chromosome (intrachromosomal) or between different chromosomes (interchromosomal). Translocations can lead to gene fusions, loss of gene function, or the misregulation of gene expression.
Copy Number Variations (CNVs) are subtypes of duplications and deletions in which the number of copies of a particular gene or genomic region varies between individuals in a population (Figure 12). CNVs can influence gene dosage expression and can be associated with various diseases and conditions.
Complex Rearrangements. These involve multiple types of SVs occurring in close proximity, often due to a single catastrophic event. Such rearrangements can include combinations of deletions, duplications, inversions, and translocations, leading to highly complex genomic alterations.
Structural variations play a significant role in human genetics and disease. They contribute to genetic diversity within populations and have been implicated in various disorders, including developmental disorders, neurological conditions, and cancers. The detection and analysis of SVs are crucial for understanding the genetic basis of disease and the study of evolution and genetic diversity. With advancements in genomic technologies, particularly high-throughput sequencing, and sophisticated computational methods, our ability to detect and characterize structural variations has improved, providing deeper insights into their roles in health and disease.
Chromosomal Alterations
Chromosomal alterations, encompassing modifications in either the structure or number of chromosomes, represent a fundamental aspect of genetic variation with significant ramifications for an organism's development, health, and evolutionary trajectory. These alterations may be inherent or acquired over the lifespan, contributing to a spectrum of genetic conditions, oncogenesis, and evolutionary diversity. They are primarily categorized into numerical and structural changes with distinct mechanisms and consequences.
Numerical chromosomal alterations pertain to variations in the chromosome count relative to the standard complement. In humans, the norm is a set of 46 chromosomes comprising 23 pairs. Deviations from this count can lead to a range of genetic conditions. Aneuploidy describes a scenario where the number of chromosomes does not conform to a complete set, manifesting as a surplus or deficit of one or more chromosomes. For example, trisomy, the presence of an extra chromosome resulting in three copies instead of the typical two, includes conditions like Down syndrome, known as Trisomy 21 (Figure 13). Conversely, monosomy, the absence of a chromosome resulting in a single copy, is exemplified by Turner syndrome, where females have a singular X chromosome. Polyploidy, a condition more prevalent in plant life and certain animal species, involves the addition of one or more complete chromosome sets, exceeding the diploid number and often leading to increased size and vitality.
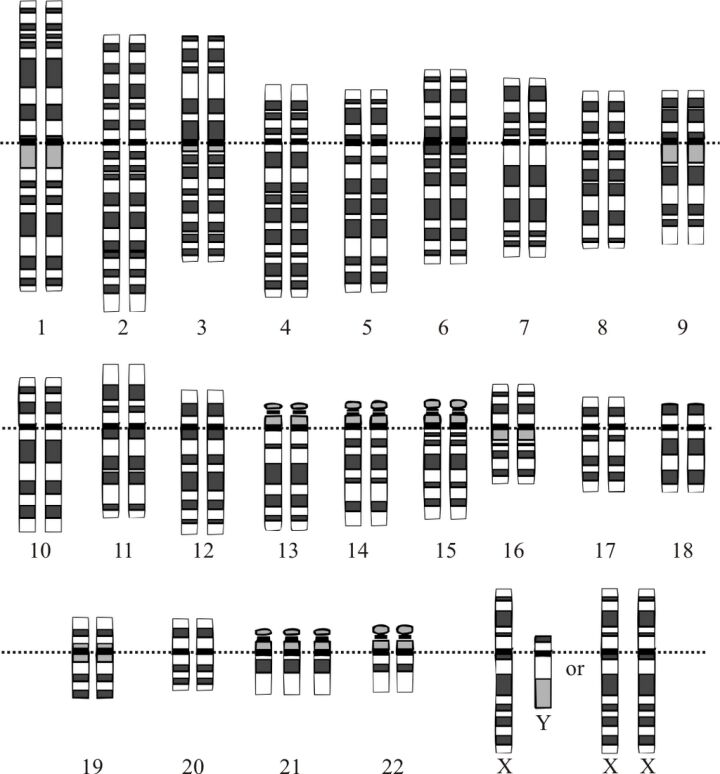
Structural alterations in chromosomes arise from modifications to the chromosome's physical architecture, often due to breaks that result in the loss, duplication, or rearrangement of genetic material. Deletions, where chromosome segments are lost, can precipitate genetic diseases by eliminating essential genes. Duplication of chromosome segments, meanwhile, introduces additional genetic material that can disrupt normal development and physiological functions. Inversions, which occur when a chromosome segment is inverted, might interfere with gene function or lead to aberrant gene regulation. Translocations entail the transfer of chromosome segments, either reciprocally between two chromosomes or non-reciprocally to another chromosome without exchange, and are linked to various cancers. Ring chromosomes, formed by the fusion of a chromosome's ends, and isochromosomes, arising from mirror-image duplication of one arm, can also result in genetic disorders due to altered genetic material distribution.
The impact of chromosomal alterations spans a broad spectrum, from negligible to severe, influencing developmental, physiological, and neurological outcomes. The specific effects depend on the implicated genes and the extent of the chromosomal changes. The detection and elucidation of these alterations are imperative for comprehending their broader implications, diagnosing genetic conditions, and guiding therapeutic development. The advent of advanced genetic and genomic technologies, such as high-resolution karyotyping, fluorescent in situ hybridization (FISH), and next-generation sequencing, has significantly enhanced our capacity to identify and characterize these chromosomal changes, marking a pivotal advancement in the field of genetics and genomics.
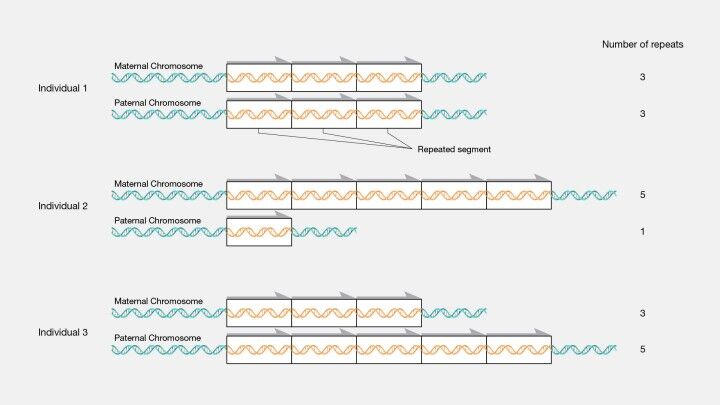
Tandem repeats
Tandem repeats constitute sequences in the DNA where nucleotides are arrayed in direct succession, creating patterns where a sequence of two or more nucleotides is repeated in a head-to-tail configuration. These sequences permeate the genome, manifesting within genes (coding regions) and intergenic spaces (non-coding regions). The spectrum of tandem repeats spans from short runs, such as microsatellites, encompassing just 2-6 base pairs (bp) (Figure 14), to the substantially longer stretches known as minisatellites or variable number tandem repeats (VNTRs), which can extend over several hundred base pairs (Figure 12).

The categorization of tandem repeats delves into their length and complexity. Microsatellites, or simple sequence repeats (SSRs), notable for their brief length of 2-6 bp, exhibit a remarkable degree of polymorphism, making them distinct across individuals within a population. This variability renders microsatellites invaluable for applications in DNA fingerprinting, the study of population genetics, and forensic analysis. On the other hand, minisatellites, or VNTRs, which are typically 10-60 bp in length, also exhibit variability among individuals and find utility in DNA fingerprinting and paternity testing. (The man behind the DNA fingerprints: an interview with Professor Sir Alec Jeffreys) Satellite DNA, comprising extensive arrays of tandem repeats, is predominantly located in heterochromatin regions such as centromeres and telomeres, playing a pivotal role in the structure and segregation of chromosomes.
The significance of tandem repeats extends into various biological functions and applications. As highly polymorphic elements, they serve as critical genetic markers in research, forensic science, and paternity tests. Specific tandem repeats situated within gene regulatory regions can influence gene expression, with the repeat count impacting transcription, translation, and protein functionality. These sequences are instrumental in promoting genomic diversity and evolution, acting as hotbeds for recombination and mutation, thereby fostering genetic variability. However, the unchecked expansion of specific tandem repeats can precipitate genomic instability and contribute to disease etiology.
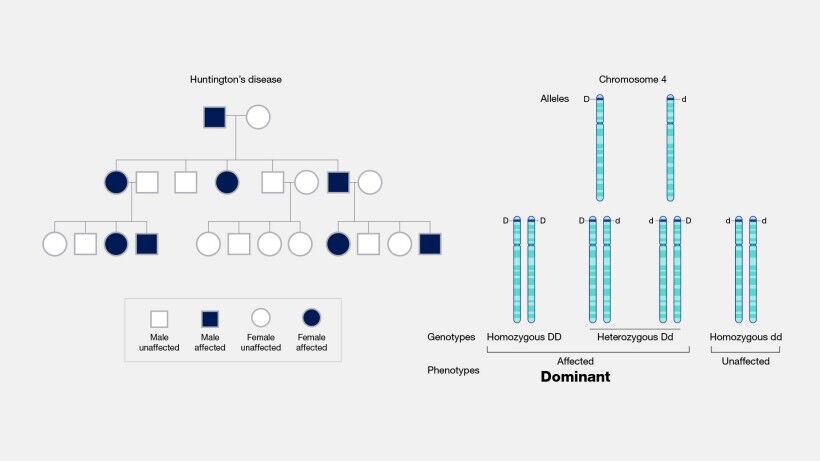
Several genetic disorders are directly linked to the expansion of tandem repeats beyond normal physiological limits. These expansions can disrupt gene function, either by encoding dysfunctional proteins or through perturbations in RNA structure and function. Notable diseases associated with tandem repeat expansions include Huntington's Disease, resulting from an extended CAG repeat in the HTT gene, which leads to neurodegenerative changes (Figure 16); Fragile X Syndrome, attributed to a CGG repeat expansion in the FMR1 gene, affecting neural development and function; Myotonic Dystrophy, characterized by a CTG repeat expansion in the DMPK gene, impacting muscle functionality and presenting a spectrum of systemic symptoms; and various forms of Spinocerebellar Ataxias, caused by CAG repeat expansions in different genes, affecting motor control.
Germline Mutations
Germline mutations are alterations that occur within an organism's reproductive cells—specifically, sperm cells in males and ovum cells in females—and have the potential to be transmitted to progeny. Such mutations are integral to the DNA of every cell in the offspring, including their germ cells, setting the stage for a hereditary chain that extends beyond a single individual to potentially influence successive generations.
The characteristics and broader implications of germline mutations are manifold, touching upon various aspects of biology, genetics, and ethics. One of these mutations' primary features is heritability; they can be passed down from parents to their offspring or might originate spontaneously in a germ cell or during the early stages of embryonic development. Once established, these mutations pervade every cell of an individual, including germ cells, thus becoming a heritable trait. This hereditary potential underscores the importance of germline mutations in the context of genetic counseling and reproductive decision-making.
From a population genetics perspective, germline mutations are a fundamental driver of genetic diversity. They introduce novel, inheritable genetic variations, playing a crucial role in evolution and shaping the genetic constitution of populations over generations. The emergence and propagation of these mutations contribute to the rich tapestry of genetic variation observed within and across species.
Germline mutations are also intimately linked with the etiology of numerous genetic disorders. Conditions such as cystic fibrosis, sickle cell anemia (Figure 15A and B), and Huntington's disease (Figure 16) can be traced back to mutations in the germline. However, it is essential to note that a germline mutation does not necessarily ensure the manifestation of the associated condition in the individual or their descendants. The expression of these mutations and their phenotypic outcomes can be influenced by various factors, including the mutation's penetrance and the interplay with other genetic and environmental factors.
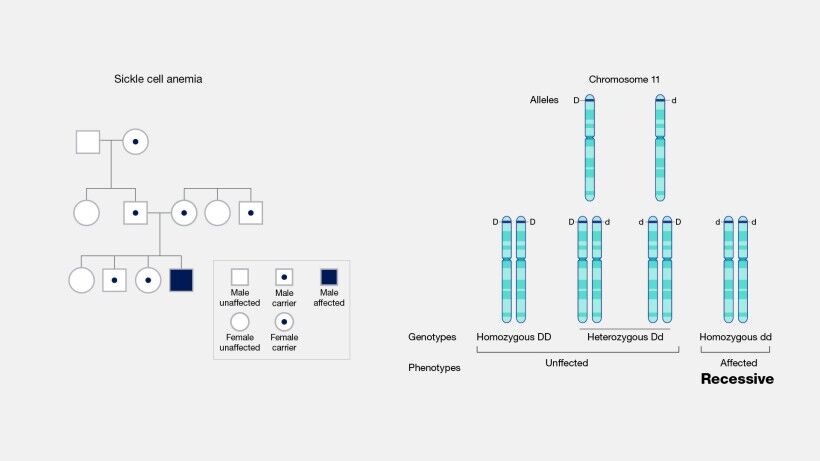
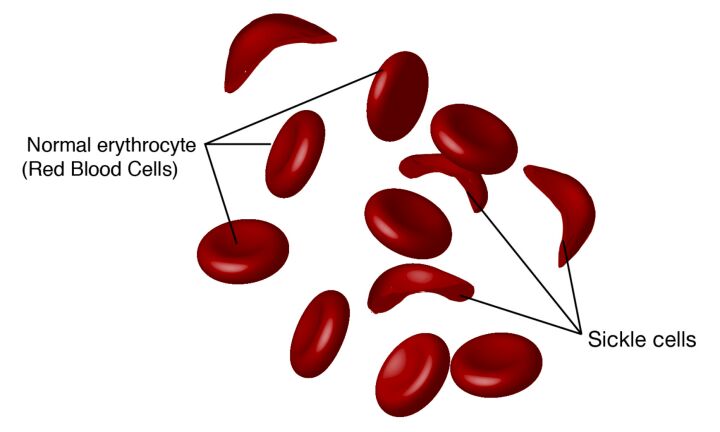
The inheritable nature of germline mutations brings significant ethical considerations, particularly in reproductive choices and genetic counseling. The potential for these mutations to be passed on to future generations necessitates informed decision-making by individuals and families who carry such mutations. Genetic counseling serves as a vital resource, offering guidance and information about the likelihood of transmitting a germline mutation to offspring, as well as the associated risks and implications for the health and well-being of future generations.
The inheritable nature of germline mutations brings significant ethical considerations, particularly in reproductive choices and genetic counseling. The potential for these mutations to be passed on to future generations necessitates informed decision-making by individuals and families who carry such mutations. Genetic counseling serves as a vital resource, offering guidance and information about the likelihood of transmitting a germline mutation to offspring, as well as the associated risks and implications for the health and well-being of future generations.
Somatic Mutations
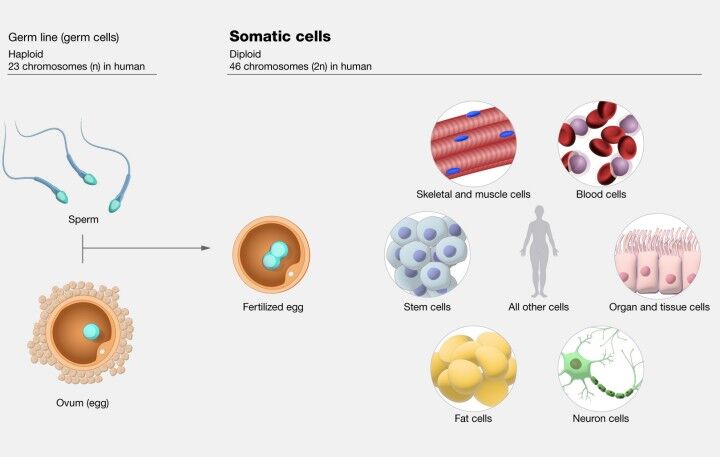
Somatic mutations represent alterations in the DNA that occur post-fertilization, manifesting in any cell type apart from the germ cells. These changes can arise at any point during an individual's lifespan, prompted by environmental factors such as ultraviolet (UV) radiation and exposure to carcinogens or as spontaneous events during DNA replication. A key characteristic of somatic mutations is their non-heritable nature; they are not transmitted to the progeny since they do not occur within germ cells.
The implications and characteristics of somatic mutations are multifaceted, impacting health and disease states in various ways. One of the fundamental aspects of somatic mutations is their clonality. A mutation originating in a single cell will propagate only through the lineage of that cell, creating a clone of cells bearing the same mutation. This concept of clonal expansion is critically relevant to the development of cancers, wherein somatic mutations drive the unregulated proliferation of cells, leading to tumor formation.
Beyond their pivotal role in oncogenesis, somatic mutations also affect other conditions, including neurodegenerative diseases. The specific impact of a somatic mutation is contingent upon the function of the gene affected and the type of cell in which the mutation occurs. Not all somatic mutations lead to cancer; their consequences vary widely, influencing disease progression and phenotype based on the nature and location of the mutation within the organism.
The phenomenon of mosaicism is another critical consideration in understanding somatic mutations. Individuals can exhibit mosaicism when a somatic mutation is present in only a subset of cells, leading to a mixed population of mutated and non-mutated cells within the body. It can result in varying degrees of disease manifestation, influenced by the distribution of mutated cells and their functional impact.
Despite their non-heritable nature, somatic mutations within an individual can indirectly influence offspring's health and disease susceptibility, highlighting the complex interplay between genetics and environmental factors in shaping health outcomes.
The delineation between germline and somatic mutations is a cornerstone of genetic understanding, with profound implications for diagnosing and treating genetic disorders, the strategic direction of cancer therapies, and insights into evolutionary biology and the development process. Grasping the nuances of somatic mutations enriches our comprehension of disease mechanisms, offering avenues for targeted interventions and therapeutic approaches in personalized medicine.
Inheritance Patterns
In the intricate tapestry of genetics, inheritance patterns play a pivotal role in determining how certain traits and mutations are transmitted from one generation to the next. These patterns, governed by the underlying genetic alterations and their localization within the genome, manifest in various forms, including dominant, recessive, X-linked, Y-linked, and mitochondrial inheritance.
Dominant mutations are characterized by their ability to express a particular phenotype with a single mutated gene copy inherited from one parent. This genetic trait implies that if an individual inherits one mutated and one normal gene copy, the effect of the mutation will be observable in the phenotype. Dominant mutations can reside on autosomes or the X chromosome, in which case they contribute to X-linked dominant conditions. Such mutations are marked by their visibility across every generation of affected families, an absence of gender bias in autosomal dominant conditions, and variability in how the mutation manifests due to differences in penetrance and expressivity.
Recessive mutations, in contrast, necessitate the inheritance of mutated gene copies from both parents for the associated phenotype to be expressed. Individuals with only one copy of the recessive mutation do not exhibit symptoms but can transmit the mutation to their offspring, serving as carriers. Recessive traits may skip generations, a phenomenon particularly noted in offspring from consanguineous unions where the chance of inheriting two copies of a recessive mutation is heightened.
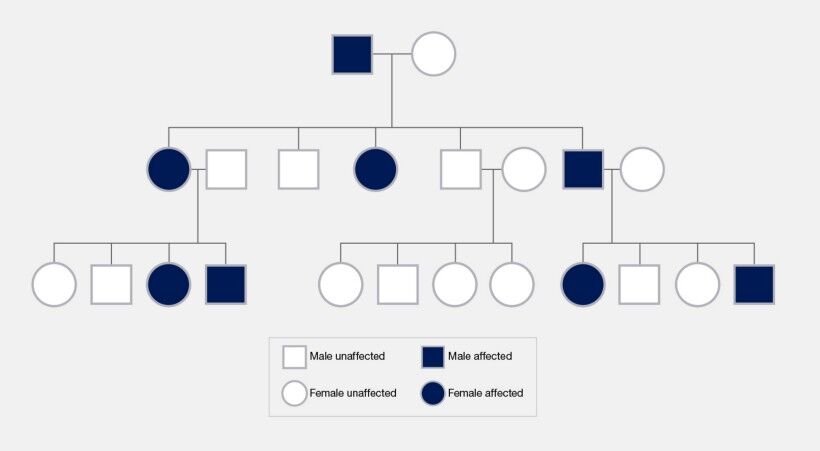
The inheritance of mutations linked to the sex chromosomes—X and Y—introduces unique patterns. X-linked mutations affect males and females differently due to the disparity in their sex chromosome composition. X-linked recessive traits affect males with a single X chromosome more frequently (Figure 17). In contrast, females possessing two X chromosomes may exhibit a mutation as carriers or be affected by X-linked dominant conditions. Y-linked mutations, transmitted from fathers to sons via the Y chromosome, are exclusive to males and influence traits tied to male development and fertility.
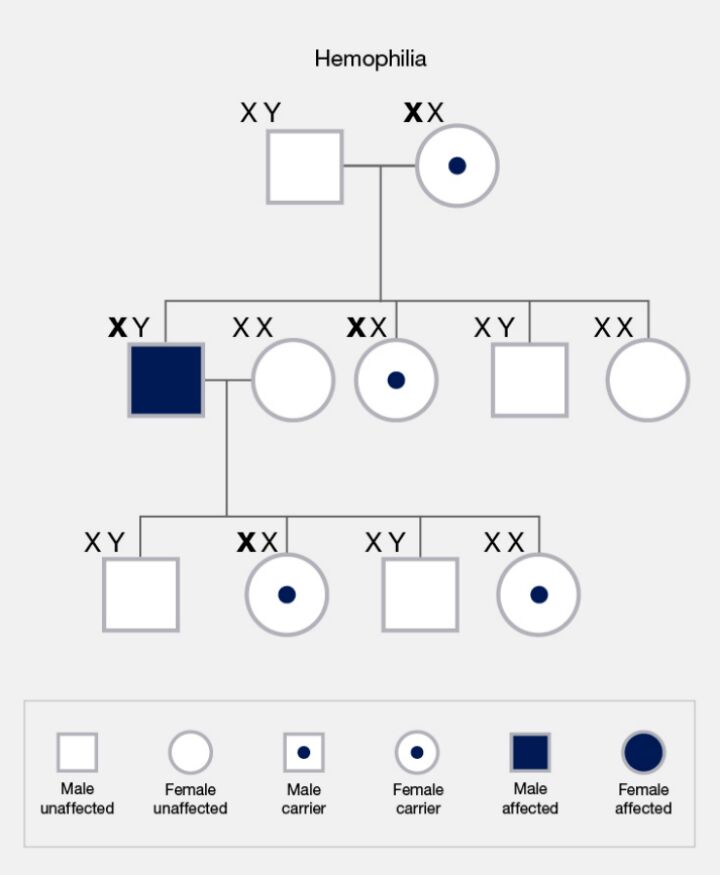
Mitochondrial inheritance adds another dimension to this genetic landscape (Figure 18). Mutations in mitochondrial DNA are transmitted from mothers to all their offspring. However, only daughters can further pass these mutations to subsequent generations, reflecting the maternal lineage of mitochondrial transmission.
Grasping the nuances of these inheritance patterns is indispensable for fields such as genetic counseling and the diagnosis of hereditary conditions. It underpins our understanding of trait and disease transmission across generations. It informs the development of management and treatment strategies for genetic disorders, enriching our comprehension of the genetic foundations that influence human health and disease.
Frequency and Distribution of Mutations
In the intricate landscape of genetics, the frequency and distribution of mutations illuminate the paths through which genetic variations influence health, disease, and evolutionary biology. This exploration delves into private mutations and common variants, each harboring unique implications for our understanding of genetic diversity and its practical applications.
Private mutations are genetic alterations peculiar to an individual, a specific family, or a small group within a population. These rare or sometimes new (de novo) mutations typically arise in a parent's germ cells or during the initial stages of embryonic development. If located in the germline, such mutations have the potential to be transmitted across generations, embedding a distinct genetic signature within a lineage. The study of private mutations is pivotal for identifying the genetic basis of rare diseases within specific families or groups, offering insights into personalized genetic counseling and shedding light on the recent evolutionary changes and genetic diversity within localized populations.
Common variants lie on the other side of the genetic variation spectrum, among which single nucleotide polymorphisms (SNPs) are predominant. SNPs represent a DNA sequence variation occurring when a single nucleotide (adenine, thymine, cytosine, or guanine) differs from the reference sequence. Typically present in at least 1% of the population, SNPs are individuals' most prevalent form of genetic variation. They may influence the risk of developing certain diseases, serve as biomarkers for predicting disease risk or drug response, or aid in tracing the inheritance of disease-causing variants within families. The minor allele frequency (MAF) of common SNPs, usually exceeding 5%, underscores their significance in the genetic constitution of populations, thereby contributing to human genetic diversity.
Minor Allele Frequency (MAF)
Minor Allele Frequency (MAF) is a term used in genetics to describe how common or rare a particular allele is within a given population. An allele is one of two or more versions of a gene, and the MAF is expressed as a proportion or percentage, representing the frequency at which the less common allele occurs in the population.
To elaborate, every individual inherits two alleles for each gene—one from each parent. In a population, there could be several alleles for a single gene, but typically one allele is more common than the others. The MAF is calculated by taking the number of times the less common allele appears in the population and dividing it by the total number of all alleles for that gene within the population.
For example, if a gene has two alleles, A and B, and allele B is present less frequently in the population than allele A, allele B is considered the minor allele. If in a population of 100 individuals (200 alleles in total for the gene, considering each individual carries two alleles), allele B is found in 40 instances, then the MAF for allele B would be 40/200, or 0.20 (20%).
Significance of MAF
- Identifying Disease Associations: Variants with a high MAF might be considered common and could be associated with common diseases or traits within the population.
- Population Genetics Studies: MAF provides insights into the genetic diversity and evolutionary patterns of populations, helping to understand how populations have adapted over time.
- Personalized Medicine: Understanding the frequency of alleles can aid in predicting disease risk and drug response in individuals, guiding more personalized healthcare strategies.
Minor Allele Frequency is a fundamental concept in genetics, offering insights into the prevalence of genetic variants within populations and playing a critical role in the study of genetics, disease association, and the development of personalized medicine approaches.
Common variants are central to population genetics research, helping elucidate populations' structure, migration patterns, and evolutionary histories. While private mutations are directly linked to rare genetic disorders, common variants reveal the multifaceted interplay between genetic predispositions and environmental factors through their association with complex traits and diseases. Identifying SNPs through genome-wide association studies (GWAS) has propelled our understanding of the genetic foundations of numerous complex conditions, paving the way for advancements in personalized medicine.
The distinction between private mutations and common variants, enriched by the detailed understanding of SNPs, captures the vast genetic variation that shapes human health, disease susceptibility, and evolutionary processes. The advancements in genomic technologies and bioinformatics are continuously enhancing our grasp of these genetic phenomena, fostering improved strategies for genetic counseling, disease prognosis, and the development of tailored medical interventions based on individual genetic profiles
Please, send
comments and suggestions. Suggest for example a topic you would like to see or any
improvements in the content. We will not spam you with emails!
References
- Elhaik, Eran. (2012). Empirical Distributions of FST from Large-Scale Human Polymorphism Data. PloS one. 7. e49837. 10.1371/journal.pone.0049837.
- Thomas Shafee; Rohan Lowe (17 January 2017). "Eukaryotic and prokaryotic gene structure". WikiJournal of Medicine 4 (1). doi:10.15347/WJM/2017.002. Wikidata Q28867140. ISSN 2002-4436.
- Lackey AE, Muzio MR. DiGeorge Syndrome. [Updated 2023 Aug 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK549798/
- Morris CA. Williams Syndrome. 1999 Apr 9 [Updated 2023 Apr 13]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1249/
- Pathak I, Bordoni B. Genetics, Chromosomes. [Updated 2023 Apr 3]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557784/
- Jiang W, Chen L. Alternative splicing: Human disease and quantitative analysis from high-throughput sequencing. Comput Struct Biotechnol J. 2020 Dec 24;19:183-195. doi: 10.1016/j.csbj.2020.12.009. PMID: 33425250; PMCID: PMC7772363.
- Anna A, Monika G. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet. 2018 Aug;59(3):253-268. doi: 10.1007/s13353-018-0444-7. Epub 2018 Apr 21. Erratum in: J Appl Genet. 2019 May;60(2):231. PMID: 29680930; PMCID: PMC6060985.
- Zhang F, Wang D. The Pattern of microRNA Binding Site Distribution. Genes (Basel). 2017 Oct 27;8(11):296. doi: 10.3390/genes8110296. PMID: 29077021; PMCID: PMC5704209.
- Macfarlane LA, Murphy PR. MicroRNA: Biogenesis, Function and Role in Cancer. Curr Genomics. 2010 Nov;11(7):537-61. doi: 10.2174/138920210793175895. PMID: 21532838; PMCID: PMC3048316.
- O'Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol (Lausanne). 2018 Aug 3;9:402. doi: 10.3389/fendo.2018.00402. PMID: 30123182; PMCID: PMC6085463.
- Zekavat, S.M., Lin, SH., Bick, A.G. et al. Hematopoietic mosaic chromosomal alterations increase the risk for diverse types of infection. Nat Med 27, 1012–1024 (2021). https://doi.org/10.1038/s41591-021-01371-0
- Brown DW, Lin SH, Loh PR, Chanock SJ, Savage SA, Machiela MJ. Genetically predicted telomere length is associated with clonal somatic copy number alterations in peripheral leukocytes. PLoS Genet. 2020 Oct 22;16(10):e1009078. doi: 10.1371/journal.pgen.1009078. PMID: 33090998; PMCID: PMC7608979.
- Thompson DJ, Genovese G, Halvardson J, Ulirsch JC, Wright DJ, Terao C, Davidsson OB, Day FR, Sulem P, Jiang Y, Danielsson M, Davies H, Dennis J, Dunlop MG, Easton DF, Fisher VA, Zink F, Houlston RS, Ingelsson M, Kar S, Kerrison ND, Kinnersley B, Kristjansson RP, Law PJ, Li R, Loveday C, Mattisson J, McCarroll SA, Murakami Y, Murray A, Olszewski P, Rychlicka-Buniowska E, Scott RA, Thorsteinsdottir U, Tomlinson I, Moghadam BT, Turnbull C, Wareham NJ, Gudbjartsson DF; International Lung Cancer Consortium (INTEGRAL-ILCCO); Breast Cancer Association Consortium; Consortium of Investigators of Modifiers of BRCA1/2; Endometrial Cancer Association Consortium; Ovarian Cancer Association Consortium; Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome (PRACTICAL) Consortium; Kidney Cancer GWAS Meta-Analysis Project; eQTLGen Consortium; Biobank-based Integrative Omics Study (BIOS) Consortium; 23andMe Research Team; Kamatani Y, Hoffmann ER, Jackson SP, Stefansson K, Auton A, Ong KK, Machiela MJ, Loh PR, Dumanski JP, Chanock SJ, Forsberg LA, Perry JRB. Genetic predisposition to mosaic Y chromosome loss in blood. Nature. 2019 Nov;575(7784):652-657. doi: 10.1038/s41586-019-1765-3. Epub 2019 Nov 20. PMID: 31748747; PMCID: PMC6887549.
- Machiela MJ, Zhou W, Karlins E, Sampson JN, Freedman ND, Yang Q, Hicks B, Dagnall C, Hautman C, Jacobs KB, Abnet CC, Aldrich MC, Amos C, Amundadottir LT, Arslan AA, Beane-Freeman LE, Berndt SI, Black A, Blot WJ, Bock CH, Bracci PM, Brinton LA, Bueno-de-Mesquita HB, Burdett L, Buring JE, Butler MA, Canzian F, Carreón T, Chaffee KG, Chang IS, Chatterjee N, Chen C, Chen C, Chen K, Chung CC, Cook LS, Crous Bou M, Cullen M, Davis FG, De Vivo I, Ding T, Doherty J, Duell EJ, Epstein CG, Fan JH, Figueroa JD, Fraumeni JF, Friedenreich CM, Fuchs CS, Gallinger S, Gao YT, Gapstur SM, Garcia-Closas M, Gaudet MM, Gaziano JM, Giles GG, Gillanders EM, Giovannucci EL, Goldin L, Goldstein AM, Haiman CA, Hallmans G, Hankinson SE, Harris CC, Henriksson R, Holly EA, Hong YC, Hoover RN, Hsiung CA, Hu N, Hu W, Hunter DJ, Hutchinson A, Jenab M, Johansen C, Khaw KT, Kim HN, Kim YH, Kim YT, Klein AP, Klein R, Koh WP, Kolonel LN, Kooperberg C, Kraft P, Krogh V, Kurtz RC, LaCroix A, Lan Q, Landi MT, Marchand LL, Li D, Liang X, Liao LM, Lin D, Liu J, Lissowska J, Lu L, Magliocco AM, Malats N, Matsuo K, McNeill LH, McWilliams RR, Melin BS, Mirabello L, Moore L, Olson SH, Orlow I, Park JY, Patiño-Garcia A, Peplonska B, Peters U, Petersen GM, Pooler L, Prescott J, Prokunina-Olsson L, Purdue MP, Qiao YL, Rajaraman P, Real FX, Riboli E, Risch HA, Rodriguez-Santiago B, Ruder AM, Savage SA, Schumacher F, Schwartz AG, Schwartz KL, Seow A, Wendy Setiawan V, Severi G, Shen H, Sheng X, Shin MH, Shu XO, Silverman DT, Spitz MR, Stevens VL, Stolzenberg-Solomon R, Stram D, Tang ZZ, Taylor PR, Teras LR, Tobias GS, Van Den Berg D, Visvanathan K, Wacholder S, Wang JC, Wang Z, Wentzensen N, Wheeler W, White E, Wiencke JK, Wolpin BM, Wong MP, Wu C, Wu T, Wu X, Wu YL, Wunder JS, Xia L, Yang HP, Yang PC, Yu K, Zanetti KA, Zeleniuch-Jacquotte A, Zheng W, Zhou B, Ziegler RG, Perez-Jurado LA, Caporaso NE, Rothman N, Tucker M, Dean MC, Yeager M, Chanock SJ. Female chromosome X mosaicism is age-related and preferentially affects the inactivated X chromosome. Nat Commun. 2016 Jun 13;7:11843. doi: 10.1038/ncomms11843. PMID: 27291797; PMCID: PMC4909985.
- Zhou W, Machiela MJ, Freedman ND, Rothman N, Malats N, Dagnall C, Caporaso N, Teras LT, Gaudet MM, Gapstur SM, Stevens VL, Jacobs KB, Sampson J, Albanes D, Weinstein S, Virtamo J, Berndt S, Hoover RN, Black A, Silverman D, Figueroa J, Garcia-Closas M, Real FX, Earl J, Marenne G, Rodriguez-Santiago B, Karagas M, Johnson A, Schwenn M, Wu X, Gu J, Ye Y, Hutchinson A, Tucker M, Perez-Jurado LA, Dean M, Yeager M, Chanock SJ. Mosaic loss of chromosome Y is associated with common variation near TCL1A. Nat Genet. 2016 May;48(5):563-8. doi: 10.1038/ng.3545. Epub 2016 Apr 11. PMID: 27064253; PMCID: PMC4848121.
- Virolainen SJ, VonHandorf A, Viel KCMF, Weirauch MT, Kottyan LC. Gene-environment interactions and their impact on human health. Genes Immun. 2023 Feb;24(1):1-11. doi: 10.1038/s41435-022-00192-6. Epub 2022 Dec 30. PMID: 36585519; PMCID: PMC9801363.
- Zou H, Wu LX, Tan L, Shang FF, Zhou HH. Significance of Single-Nucleotide Variants in Long Intergenic Non-protein Coding RNAs. Front Cell Dev Biol. 2020 May 25;8:347. doi: 10.3389/fcell.2020.00347. PMID: 32523949; PMCID: PMC7261909.
- Pös, O., Radvanszky, J., Buglyó, G., Pös, Z., Rusnakova, D., Nagy, B., & Szemes, T. (2021). DNA copy number variation: Main characteristics, evolutionary significance, and pathological aspects. Biomedical Journal, 44(5), 548-559. https://doi.org/10.1016/j.bj.2021.02.003
- Ameen, S.K., Alalaf, S.K. & Shabila, N.P. Pattern of congenital anomalies at birth and their correlations with maternal characteristics in the maternity teaching hospital, Erbil city, Iraq. BMC Pregnancy Childbirth 18, 501 (2018). https://doi.org/10.1186/s12884-018-2141-2
- Bell JI. Single nucleotide polymorphisms and disease gene mapping. Arthritis Res. 2002;4 Suppl 3(Suppl 3):S273-8. doi: 10.1186/ar555. Epub 2002 May 9. PMID: 12110147; PMCID: PMC3240131.
- Brown TA. Genomes. 2nd edition. Oxford: Wiley-Liss; 2002. Chapter 14, Mutation, Repair and Recombination. Available from: https://www.ncbi.nlm.nih.gov/books/NBK21114/
- Balachandran P, Beck CR. Structural variant identification and characterization. Chromosome Res. 2020 Mar;28(1):31-47. doi: 10.1007/s10577-019-09623-z. Epub 2020 Jan 6. PMID: 31907725; PMCID: PMC7131885.
- Karaoğlanoğlu, F., Ricketts, C., Ebren, E. et al. VALOR2: characterization of large-scale structural variants using linked-reads. Genome Biol 21, 72 (2020). https://doi.org/10.1186/s13059-020-01975-8
- Yang L. A Practical Guide for Structural Variation Detection in the Human Genome. Curr Protoc Hum Genet. 2020 Sep;107(1):e103. doi: 10.1002/cphg.103. PMID: 32813322; PMCID: PMC7738216.
- Cui X, Cui Y, Shi L, Luan J, Zhou X, Han J. A basic understanding of Turner syndrome: Incidence, complications, diagnosis, and treatment. Intractable Rare Dis Res. 2018 Nov;7(4):223-228. doi: 10.5582/irdr.2017.01056. PMID: 30560013; PMCID: PMC6290843.
- Morgan T. Turner syndrome: diagnosis and management. Am Fam Physician. 2007 Aug 1;76(3):405-10. PMID: 17708142.
- Yip MY. Autosomal ring chromosomes in human genetic disorders. Transl Pediatr. 2015 Apr;4(2):164-74. doi: 10.3978/j.issn.2224-4336.2015.03.04. PMID: 26835370; PMCID: PMC4729093.
- Li P, Dupont B, Hu Q, Crimi M, Shen Y, Lebedev I, Liehr T. The past, present, and future for constitutional ring chromosomes: A report of the international consortium for human ring chromosomes. HGG Adv. 2022 Sep 10;3(4):100139. doi: 10.1016/j.xhgg.2022.100139. PMID: 36187226; PMCID: PMC9519620.
- Nikitina, T.V., Kashevarova, A.A., Gridina, M.M. et al. Complex biology of constitutional ring chromosomes structure and (in)stability revealed by somatic cell reprogramming. Sci Rep 11, 4325 (2021). https://doi.org/10.1038/s41598-021-83399-3
- Mao X, James SY, Yáñez-Muñoz RJ, Chaplin T, Molloy G, Oliver RT, Young BD, Lu YJ. Rapid high-resolution karyotyping with precise identification of chromosome breakpoints. Genes Chromosomes Cancer. 2007 Jul;46(7):675-83. doi: 10.1002/gcc.20452. PMID: 17431877.
- Slater HR, Bailey DK, Ren H, Cao M, Bell K, Nasioulas S, Henke R, Choo KH, Kennedy GC. High-resolution identification of chromosomal abnormalities using oligonucleotide arrays containing 116,204 SNPs. Am J Hum Genet. 2005 Nov;77(5):709-26. doi: 10.1086/497343. Epub 2005 Sep 16. Erratum in: Am J Hum Genet. 2006 Mar;78(3):526. PMID: 16252233; PMCID: PMC1271402.
- Shakoori AR. Fluorescence In Situ Hybridization (FISH) and Its Applications. Chromosome Structure and Aberrations. 2017 Feb 10:343–67. doi: 10.1007/978-81-322-3673-3_16. PMCID: PMC7122835.
- Behjati S, Tarpey PS. What is next generation sequencing? Arch Dis Child Educ Pract Ed. 2013 Dec;98(6):236-8. doi: 10.1136/archdischild-2013-304340. Epub 2013 Aug 28. PMID: 23986538; PMCID: PMC3841808.
- Sinclair A. Genetics 101: cytogenetics and FISH. CMAJ. 2002 Aug 20;167(4):373-4. PMID: 12197695; PMCID: PMC117855.
- Roewer L. DNA fingerprinting in forensics: past, present, future. Investig Genet. 2013 Nov 18;4(1):22. doi: 10.1186/2041-2223-4-22. PMID: 24245688; PMCID: PMC3831584.
- Jeffreys AJ. The man behind the DNA fingerprints: an interview with Professor Sir Alec Jeffreys. Investig Genet. 2013 Nov 18;4(1):21. doi: 10.1186/2041-2223-4-21. PMID: 24245655; PMCID: PMC3831583.
- Thakur J, Packiaraj J, Henikoff S. Sequence, Chromatin and Evolution of Satellite DNA. Int J Mol Sci. 2021 Apr 21;22(9):4309. doi: 10.3390/ijms22094309. PMID: 33919233; PMCID: PMC8122249.
- Nopoulos PC. Huntington disease: a single-gene degenerative disorder of the striatum. Dialogues Clin Neurosci. 2016 Mar;18(1):91-8. doi: 10.31887/DCNS.2016.18.1/pnopoulos. PMID: 27069383; PMCID: PMC4826775.
- Protic DD, Aishworiya R, Salcedo-Arellano MJ, Tang SJ, Milisavljevic J, Mitrovic F, Hagerman RJ, Budimirovic DB. Fragile X Syndrome: From Molecular Aspect to Clinical Treatment. Int J Mol Sci. 2022 Feb 9;23(4):1935. doi: 10.3390/ijms23041935. PMID: 35216055; PMCID: PMC8875233.
- Peterson JAM, Cooper TA. Clinical and Molecular Insights into Gastrointestinal Dysfunction in Myotonic Dystrophy Types 1 & 2. Int J Mol Sci. 2022 Nov 26;23(23):14779. doi: 10.3390/ijms232314779. PMID: 36499107; PMCID: PMC9737721.
- Sznajder ŁJ, Swanson MS. Short Tandem Repeat Expansions and RNA-Mediated Pathogenesis in Myotonic Dystrophy. Int J Mol Sci. 2019 Jul 9;20(13):3365. doi: 10.3390/ijms20133365. PMID: 31323950; PMCID: PMC6651174.
- Soltanzadeh P. Myotonic Dystrophies: A Genetic Overview. Genes (Basel). 2022 Feb 17;13(2):367. doi: 10.3390/genes13020367. PMID: 35205411; PMCID: PMC8872148.
- Buijsen RAM, Toonen LJA, Gardiner SL, van Roon-Mom WMC. Genetics, Mechanisms, and Therapeutic Progress in Polyglutamine Spinocerebellar Ataxias. Neurotherapeutics. 2019 Apr;16(2):263-286. doi: 10.1007/s13311-018-00696-y. PMID: 30607747; PMCID: PMC6554265.
- Ashizawa T, Öz G, Paulson HL. Spinocerebellar ataxias: prospects and challenges for therapy development. Nat Rev Neurol. 2018 Oct;14(10):590-605. doi: 10.1038/s41582-018-0051-6. Erratum in: Nat Rev Neurol. 2018 Dec;14(12):749. PMID: 30131520; PMCID: PMC6469934.
- Polonsky M, Chain B, Friedman N. Clonal expansion under the microscope: studying lymphocyte activation and differentiation using live-cell imaging. Immunol Cell Biol. 2016 Mar;94(3):242-9. doi: 10.1038/icb.2015.104. Epub 2015 Dec 22. PMID: 26606974.
- Adams NM, Grassmann S, Sun JC. Clonal expansion of innate and adaptive lymphocytes. Nat Rev Immunol. 2020 Nov;20(11):694-707. doi: 10.1038/s41577-020-0307-4. Epub 2020 May 18. PMID: 32424244.
- Evans MA, Walsh K. Clonal hematopoiesis, somatic mosaicism, and age-associated disease. Physiol Rev. 2023 Jan 1;103(1):649-716. doi: 10.1152/physrev.00004.2022. Epub 2022 Sep 1. PMID: 36049115; PMCID: PMC9639777.
- Park SJ, Bejar R. Clonal hematopoiesis in cancer. Exp Hematol. 2020 Mar;83:105-112. doi: 10.1016/j.exphem.2020.02.001. Epub 2020 Feb 7. PMID: 32044376; PMCID: PMC7103485.
- Lee, M., Lui, A.C.Y., Chan, J.C.K. et al. Revealing parental mosaicism: the hidden answer to the recurrence of apparent de novo variants. Hum Genomics 17, 91 (2023). https://doi.org/10.1186/s40246-023-00535-y